Never Miss a Reportable Event. Ever.
Smarteeva's AI-powered adverse event management detects potential reportable events from complaint narratives automatically, generates regulatory submissions in minutes, and provides complete tracking of MDRs, MIRs, CVRs, PSURs, and global adverse event reports, with smart audit trail and e-signatures built in.
- AI-Powered Adverse Event Detection
- Automated Regulatory Report Generation
- Global Regulatory Framework Support








Core capabilities
AI-Powered Adverse Event Detection
Smarteeva's NLP engine processes every complaint narrative in your system, every word, every sentence, every time. It doesn't rely on simple keyword matching. The AI understands clinical context, recognizes indirect references to patient harm or device malfunction, and identifies language patterns that indicate reportable conditions even when the complaint doesn't use standard regulatory terminology.
When a potential adverse event is detected, the system generates an alert that includes: the specific text that triggered the detection, the classified event type (injury, malfunction, death), the applicable regulatory framework (FDA MDR, EU Vigilance, etc.), and a confidence score. Your regulatory team reviews the flagged event with complete context - not a vague notification that says "please review this complaint."
Automated Regulatory Report Generation
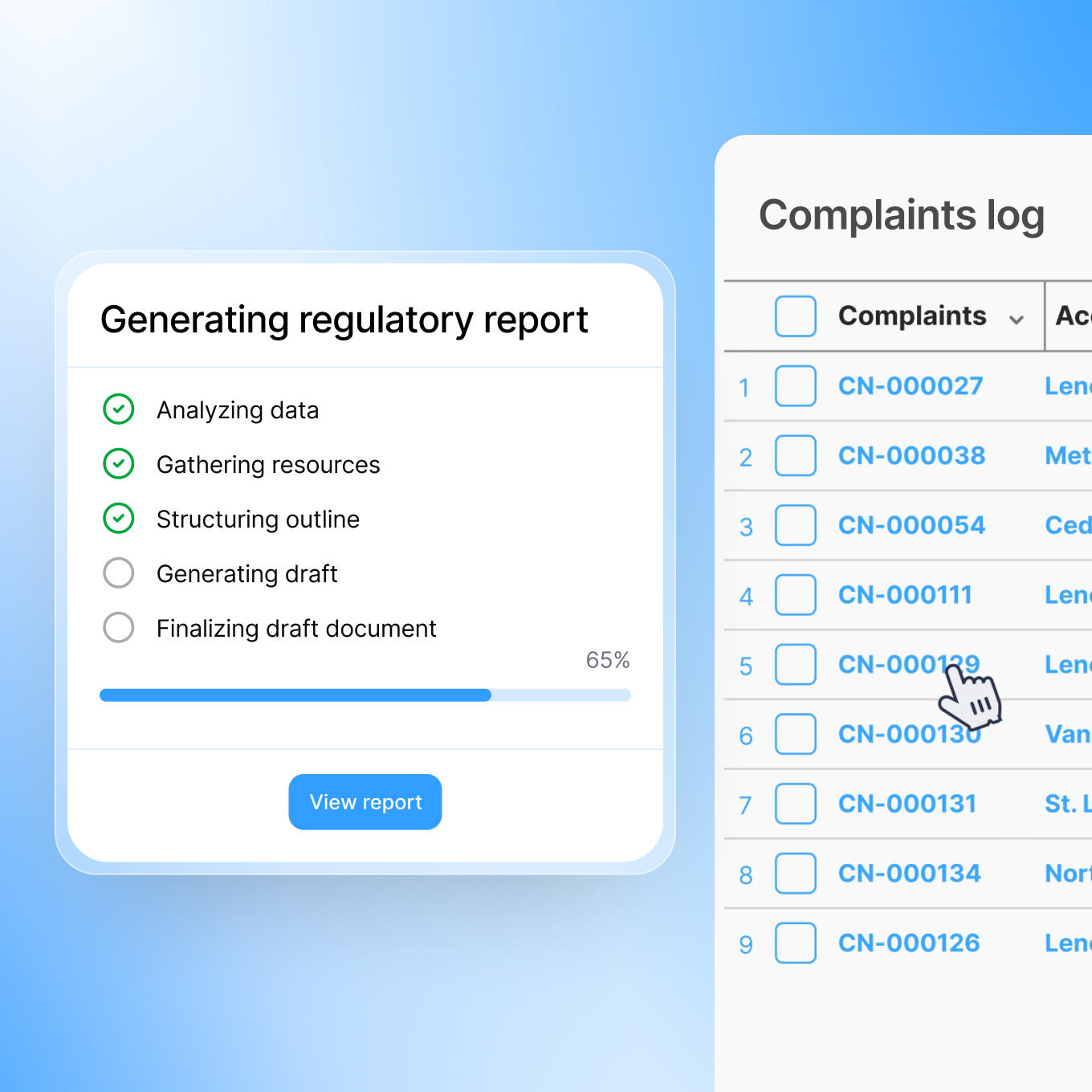
Once your team confirms a reportable event, Smarteeva generates the regulatory submission automatically. For MedWatch/MDR reports, the system maps complaint data, adverse event assessment data, product information, and patient outcomes to the required reporting fields. Draft reports are produced in minutes, complete, properly formatted, and ready for your regulatory team's expert review.
Before submission, Smarteeva validates data integrity. Missing fields are flagged. Inconsistencies are surfaced. Your team catches issues before filing, not after an agency follow-up.
Global Regulatory Framework Support
Adverse event reporting isn't a single-market obligation. If you sell medical devices globally, you report to multiple agencies with different requirements, different forms, and different timelines. Smarteeva supports:
- FDA — MedWatch / MDR (Medical Device Reports)
- EU — Vigilance Reporting under EU MDR (2017/745)
- UK — Vigilance under UK MDR
- Japan — PMDA adverse event reporting
- Australia — TGA adverse event reporting
- Additional frameworks — MIRs, CVRs, PSURs, and other region-specific reports
E-Signatures & Submission Approval
Regulatory submissions require formal approval before filing. Smarteeva's built-in e-signature capability, inherited from the Salesforce platform, allows your regulatory team to review, approve, and sign off on submissions electronically, with full audit trail documentation of who approved what and when.
Multi-Device Access
Your regulatory team doesn't always work at a desk. Smarteeva's adverse event management is accessible from any device, desktop, laptop, tablet - so your team can review flagged events, approve submissions, and manage reporting deadlines from wherever they work.
Regulatory Compliance at Stake
Global Regulatory Demands
AI That Ensures Nothing Is Missed
The regulated industries that depend on us.