
Stop Assembling Regulatory Submissions. Start Generating Them.
Smarteeva automates MedWatch reports, MDR filings, EU PSURs, PMSRs, and global adverse event submissions, mapping your quality data to regulatory fields and producing submission-ready documents in minutes. Your regulatory team's expertise belongs in the analysis, not the data assembly.







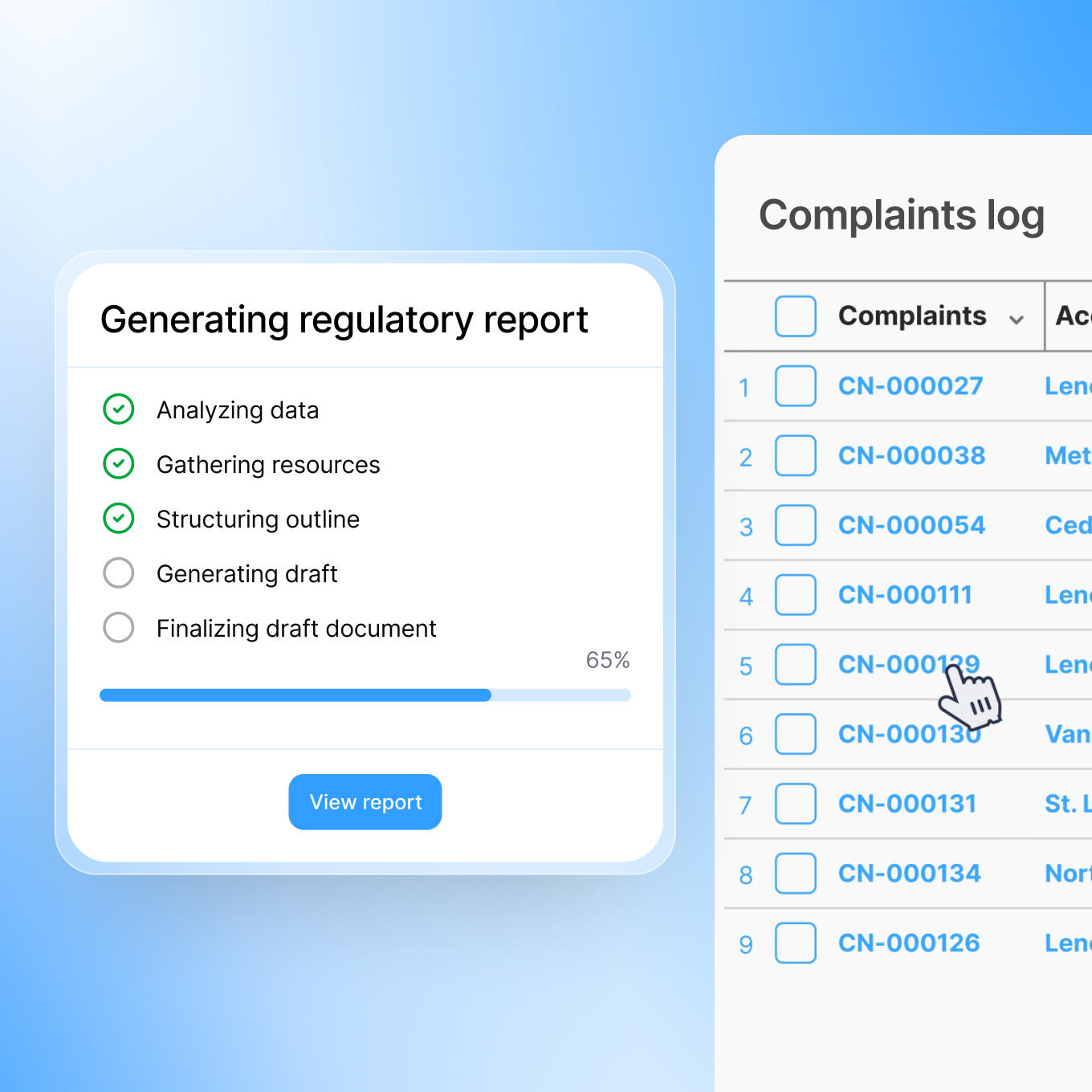
That's why we built a platform that generates regulatory submissions for you, automatically, completely, and on time.
FDA MedWatch / MDR Reports
When an adverse event is confirmed as reportable, Smarteeva generates a draft MedWatch report automatically. Complaint data, adverse event assessment data, product information, patient outcome details, and investigation findings are mapped to the appropriate MedWatch form fields. The draft includes pre-populated narrative sections based on the complaint record, ready for your regulatory team to refine and finalize.
Before the report is submitted, Smarteeva runs data validation checks: missing required fields are flagged, inconsistencies between the complaint record and the submission are highlighted, and completeness is verified. Your team catches errors before filing, not after an FDA follow-up request.
EU PSUR (Periodic Safety Update Report)
EU MDR Article 86 requires Periodic Safety Update Reports for Class IIa, IIb, and III devices. These are among the most data-intensive regulatory documents in post-market surveillance, requiring complaint summaries, adverse event analyses, trend data, risk-benefit assessments, and conclusions across entire product families and multi-year reporting periods.
Smarteeva generates complete PSUR documents in 8 minutes by pulling complaint data, adverse event records, and quality data for the specified product family and period. Statistical summaries, trend analyses, and regulatory-formatted sections are assembled automatically into an audit-ready document.
EU PMSR (Post-Market Surveillance Report)
For Class I devices, EU MDR Article 85 requires Post-Market Surveillance Reports. Smarteeva generates PMSRs by compiling PMS activities, complaint data, corrective actions, and conclusions on the need for preventive measures.
UK PSUR & UK PMSR
Smarteeva generates the UK MDR equivalents with regulatory alignment specific to UK requirements.
PMPF-ER (Post-Market Performance Follow-up Evaluation Report)
For IVD manufacturers under the EU IVDR, Smarteeva compiles performance data, complaint trends, and clinical evidence into the required evaluation report format.
HCSR (Health Canada Summary Report)
Aligned with Health Canada's post-market surveillance reporting requirements, Smarteeva generates HCSRs from your existing quality data.
Global Adverse Event Reports
Beyond MedWatch, Smarteeva supports the creation of adverse event reports for EU Vigilance, UK Vigilance, PMDA (Japan), TGA (Australia), and other jurisdictions, mapping your complaint data to each framework's specific requirements.
How Automated Submission Generation Works
Step 1: Trigger
A reportable event is confirmed, a PMS report period closes, or a manual request initiates the report generation process
Step 2: Data Pull
Smarteeva's AI gathers all relevant complaint records, adverse event data, investigation outcomes, product information, and quality data for the specified scope and period. No manual data gathering.
Step 3: Mapping & Assembly
Data is mapped to the required regulatory form fields and report sections. For narrative-based reports (PSURs, PMSRs), the AI computes statistical summaries, generates trend analyses, and assembles the regulatory-required sections.
Step 4: Validation
The system runs completeness and consistency checks. Missing fields, data conflicts, and formatting issues are flagged before anyone reviews the document.
Step 5: Expert Review
Your regulatory team receives a complete, formatted draft document. They review the content, add expert commentary, refine narrative descriptions, ensure conclusions are accurate, and approve for submission.
Step 6: Submission & Audit Trail
The finalized report is submitted with e-signatures and a complete audit trail documenting who generated, reviewed, approved, and submitted the document.
From Weeks to Minutes.
For a company filing 50+ MedWatch reports per year and producing PSURs for multiple product families, the annualized time savings often exceed thousands of hours, freeing your regulatory team to focus on submission quality, agency interactions, and strategic regulatory planning.